ANNALS D'OFTALMOLOGIA
DL. (v. electrónica):
B-16965-2010
ISSN (v. electrónica):
2013-8415
4 números/año
INSCRÍBETE A LA NEWSLETTER

Volumen 22 - Número 4 - Octubre - Noviembre 2014
Rabdomiosarcoma orbitario primario: hallazgo tras traumatismo ocular accidental
C. Sierra Alonso1, E. Casas Gimeno2
1Hospital de l’Esperança. Parc de Salut Mar. Barcelona
2Hospital Sant Joan de Déu. Esplugues de Llobregat. Barcelona.
2Hospital Sant Joan de Déu. Esplugues de Llobregat. Barcelona.
CORRESPONDENCIA
Carlos Sierra Alonso
E-mail: CSierra@parcdesalutmar.cat
E-mail: CSierra@parcdesalutmar.cat
RESUMEN
El rabdomiosarcoma es una neoplasia infrecuente con una incidencia baja, pero representa desde el punto de vista oftalmológico, el tumor maligno orbitario más común en la edad pediátrica que compromete la vida del paciente. Presentamos el caso clínico de un varón cuyo hallazgo se realizó tras un traumatismo orbitario accidental. Es clave conocer la presentación clínica, subtipos histológicos, clasificación y características inmunohistoquímicas y genéticas de este tipo de tumoración para poder hacer un diagnóstico temprano, ofrecer un manejo multidisciplinario y un pronóstico aproximado. Los nuevos regímenes quimioterápicos actuales están aumentando radicalmente las tasas de supervivencia del rabdomiosarcoma.
RESUM
El rabdomiosarcoma és una neoplàsia infreqüent, amb una baixa incidència, però que representa des del punt de vista oftalmològic, el tumor maligne orbitari més comú a l’edat pediàtrica que compromet la vida del pacient. Presentem el cas clínic d’un varó on la troballa es va fer arrel d’un traumatisme orbitari accidental. És clau conèixer la presentació clínica, subtipus histològics, classificació i característiques inmunohistoquímiques i genètiques d’aquest tipus de tumoració per poder fer un diagnòstic d’hora, oferir un maneig multidisciplinari i una aproximació pronòstica. Els nous règims quimioteràpics actuals estan augmentant de manera radical les tasses de supervivència del rabdomiosarcoma.
ABSTRACT
Rhabdomyosarcoma is a rare neoplasm with a low incidence but it is ophthalmologically the most common malignant orbital tumor in children that involves the patient's life. We report the case of a man whose discovery was made after an accidental orbital trauma. It is essential to know the clinical presentation, histological subtypes, classification and immunohistochemical and genetic characteristics of this type of tumor to make an early diagnosis, provide a multidisciplinary management and an approximate prognosis. The new current chemotherapy regimens are dramatically increasing survival rates of rhabdomyosarcoma.
Introducción
El rabdomiosarcoma (RMS), del griego ῥαβδω rhabdo- (estriado) μυς myo- (músculo) y sarkos (tumor carnoso, embriológicamente derivado del mesodermo), es el sarcoma de partes blandas más frecuente en la edad pediátrica, que forma parte de hasta un 5% de todas las neoplasias malignas infantiles y un 20% de todos los tumores de partes blandas.Webner fue el primero en describir un caso de RMS lingual en 1854. Bayer ofreció la primera descripción del RMS orbitario en el 1882. La definición histopatológica no llegó hasta 1946.
La mitad de rabdomiosarcomas se presentan a nivel de cabeza y cuello, y el resto se reparte en tronco y extremidades (la localización más frecuente en adultos). Puede aparecer en cualquier localización anatómica donde haya músculo esquelético o no; como la bufeta o los conductos biliares.
El rabdomiosacorma orbitario primario representa el 25-35% de los rabdomiosarcomas de cabeza y cuello. Se presenta característicamente en niños más que en niñas (ratio 5:3), a una edad media de 8 años, de manera unilateral y sin preferencia racial1,2.
Aunque el rabdomiosarcoma primario puede aparecer en otras zonas como conjuntiva, párpados, e incluso en úvea; la órbita, es la región preferente en la localización de este tipo de tumores.
A continuación, presentamos el caso clínico de un RMS orbitario primario cuya detección se realizó tras un traumatismo orbitario accidental, retrasando el diagnóstico hasta su hallazgo intraquirúrgico. Se han descrito algunos casos precedentes de RMS orbitario primario tras un traumatismo de órbita, que han demorado el diagnóstico definitivo3,4.
Caso clínico
Varón de doce años de edad sin antecedentes patológicos ni oftalmológicos que acudió al servicio de urgencias por diplopía binocular de predominio a la mirada a la izquierda de una semana de evolución. Refería traumatismo (golpe de puño) en ojo izquierdo ocho días antes.En la exploración física destacaba una agudeza de 1.0 en ambos ojos, diplopía vertical, déficit en la supraducción y levoversión, hipoftalmos marcado con ausencia de restricción inferior. Se palpaba una lesión a nivel superomedial de la órbita izquierda y retropulsión positiva del globo ocular.
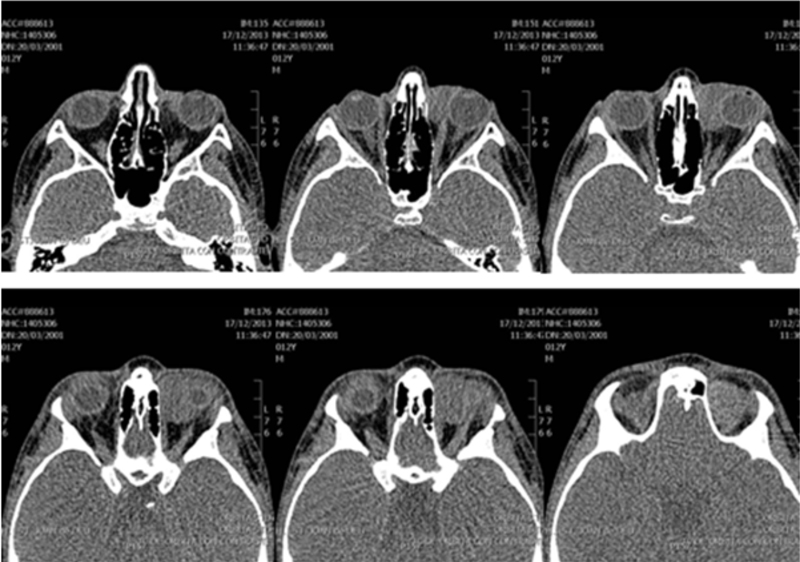
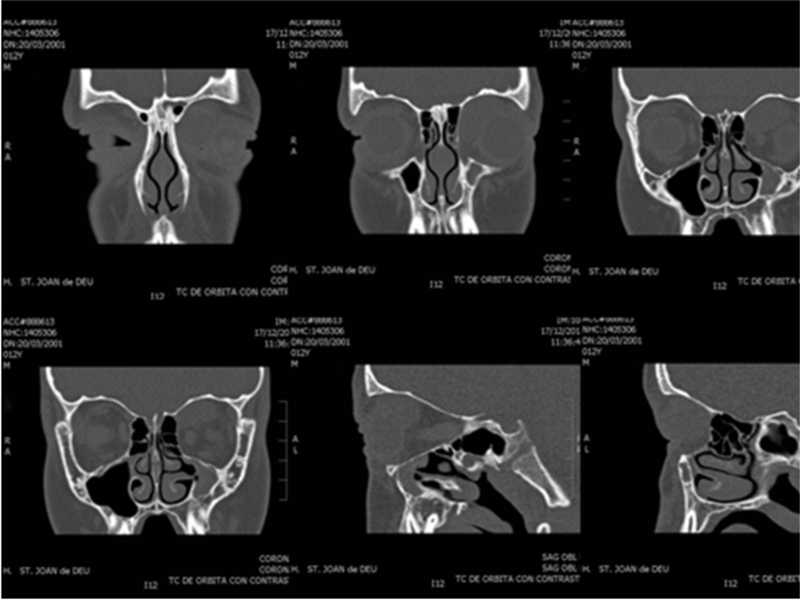
La tomografía computerizada (TC) de órbita (Figura 1 y Figura 2) evidenciaba una lesión ocupante de espacio de densidad de partes blandas de 3 centímetros de diámetro localizada a nivel superomedial de la órbita izquierda y que desplazaba el globo ocular anterolateralmente, así como al recto interno y el recto-oblicuo superior. No se apreciaban lesiones óseas, ni afectación de senos paranasales ni frontales.
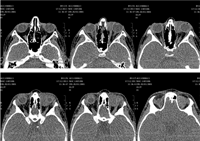
Figura 1. TC órbita (axial).
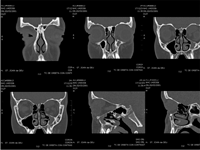
Figura 2. TC órbita (coronal y sagital de órbita izquierda).
El diagnóstico de sospecha, en este momento, fue de hematoma postraumático, sangrado sobre proceso linfangiomatoso o tumor primario orbitario.
Se procedió a realización de orbitotomía antero-supero-medial izquierda para drenaje de posible hematoma y exploración in situ de la lesión, evidenciando una lesión de tipo tumoral, blanca y semidura a la palpación compatible con rabdomiosarcoma orbitario que fue confirmada por la histología e inmunohistoquímica de anatomía patológica.
Se realizó PCR en busca de la expresión de las translocaciones características de RMS alveolar: PAX3-FKHR y PAX7-FKHR que fueron negativas, con una amplificación correcta del gen control.
Se contactó con el servicio de oncología para estudio de extensión e inicio de tratamiento quimioterápico. Tras la realización de TC toraco-abdominal y aspirado y biopsia de médula ósea, sin hallazgos de diseminación a distancia del proceso de base se cataloga de rabdomiosarcoma embrionario primario orbitario localizado y se inicia quimioterapia según Protocolo EpSSG (European Paediatric Soft Tissue Sarcoma Study Group) para rabdomiosarcoma riesgo estándar (subgrupo C).
Histopatología
A nivel histopatológico, el rabdomiosarcoma se clasifica como una neoplasia maligna derivada de células de músculo estriado en diversos estados embrionarios. Se cree que su origen se debe a células mesenquimales pluripotenciales con tendencia a diferenciarse a células musculo-esqueléticas, de manera que su origen no precisa de una localización musculoesquelética. (Puede aparecer en cualquier localización, excepto en hueso) .Se distinguen cuatro subtipos histológicos: embrionario, alveolar, pleomórfico y botriode.
- Embrionario (80%). El más frecuente y de mejor pronóstico (94% supervivencia a los 5 años). Localización: cabeza, cuello y órbita. La histología muestra células fusiformes a redondas que muestran características de músculo estriado en varios estadios de desarrollo embrionario, de citoplasma eosinófilo, y presencia de estriaciones cruzadas (Figura 3).
- Alveolar. El menos frecuente y de peor pronóstico. 74% supervivencia a los 5 años)
– Células lisas con septos que recuerdan a los alvéolos pulmonares.
- Botriode.
– Configuración papilar.
- Pleomórfico: Muy raro en la órbita, y más frecuente en adultos.
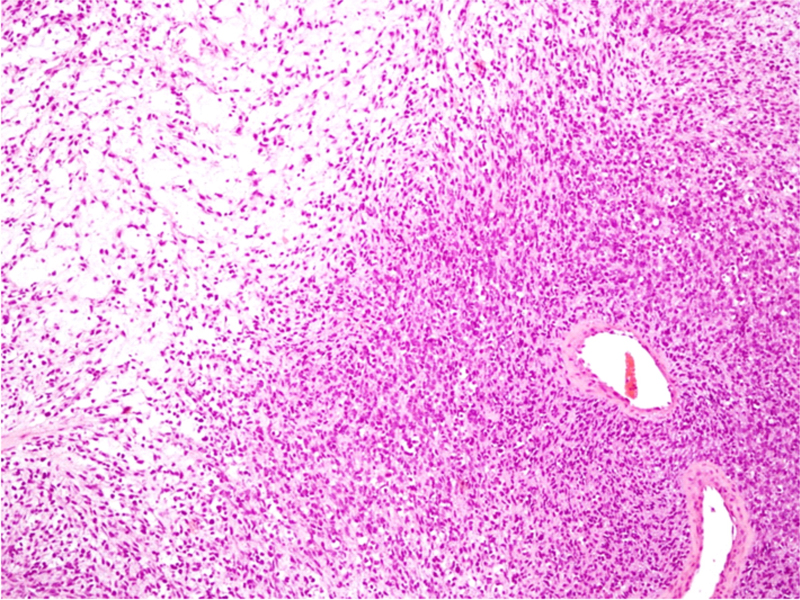
Figura 3. Histología de rabdomiosarcoma embrionario (tinción H/E): Tumoración constituida por una proliferación de células fusiformes, densamente celulares, agrupadas alrededor de estructuras vasculares y con zonas de fondo claramente mixoide. Las células tumorales muestran núcleos elongados de cromatina fina, pequeño nucléolo y citoplasmas mal delimitados, con abundantes figuras de mitosis.
Las pruebas de inmunohistoquímica se utilizan como ayuda diagnóstica y algunos parámetros podrían ser claves pronósticas5: anticuerpos anti-desmina, actina músculo-específica, mioglobina, vimentina (es menos específica) (Figura 4).
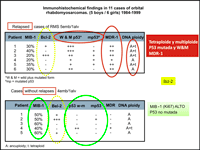
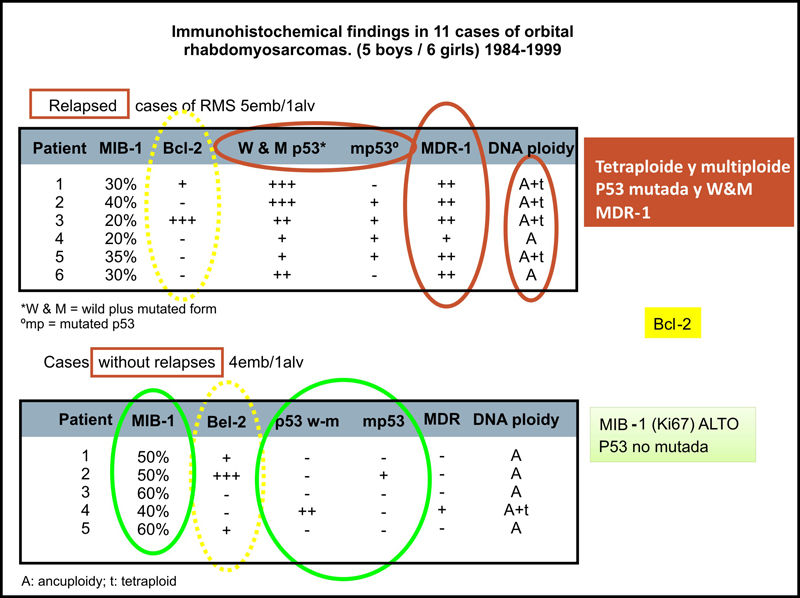
Figura 4. En este estudio se relacionaron varias variables inmunohistoquímicas con su relación pronóstica, en base a la aparición o no de recidiva tumoral. En rojo: variables de mal pronóstico. En verde: variables de buen pronóstico. Amarillo: sin significación estadística.
El RMS alveolar se asocia en un 80-85% a unas translocaciones específicas entre los brazos largos del cromosoma 2 y cromosoma 13, que se conoce como t (2:13) (q35;q14). Esta translocación fusiona el gen PAX3 (se cree que regula la transcripción durante el desarrollo neuromuscular temprano) con el gen FHKR (miembro de una familia de factores de transcripción conocida como forkhead family of transcription factors). Se cree que esta fusión anómala activa inapropiadamente los genes de transcripción hacia un determinado fenotipo.
La variante t (1:13) (p26;q14) fusiona el gen PAX7 localizado con el cromosoma 1 con el gen FHKR.
Los pacientes que expresan la fusión PAX-FKHR tienden a ser pacientes más jóvenes y frecuentemente presentan tumoración en una de las extremidades. La amplificación por PCR de estos genes es ahora también un método diagnóstico al alcance, que permite la confirmación del diagnóstico de RMS subtipo alveolar6.
Discusión
El rabdomiosarcoma precisa de un reconocimiento oftalmológico temprano y por ello debe ser siempre sospechado ante un niño entre la primera y segunda década de la vida (media de 6 a 8 años) que se presenta con proptosis y desplazamiento del globo ocular de rápida evolución, preferentemente inferotemporal, pues la localización del tumor en la órbita es en un 70% superior y superonasal, y aproximadamente la mitad se localiza en la órbita media1,2.Puede haber presentes otros signos y síntomas:
- 80-100% proptosis
- 80% desplazamiento del globo ocular
- 60% edema palpebral y conjuntival
- 30-50% blefaroptosis (a veces primer signo de un tumor orbitario superior)
- 25% masa palpable
- 10% dolor
En el fondo de ojo se pueden hallar: pliegues coroideos, tortuosidad venosa y edema del disco óptico.
Los hallazgos radiológicos típicos se caracterizan por una masa circunscrita, homogénea, de redonda a oval, isodensa, usualmente extraconal, de localización superonasal, confinada a tejidos blandos, (no procede de la musculatura) que puede mostrar desplazamiento de MOEs sin alargamiento del vientre muscular, con moderado a marcado realce con contraste y extensión orbitaria ósea (tardía).
Se debe realizar el diagnóstico diferencial con toda una serie de enfermedades que pueden afectar la órbita (Tabla 1). En nuestro caso, el antecedente de traumatismo orbitario reciente previo, dificulta el diagnóstico de sospecha y requiere realizar un diagnóstico diferencial, principalmente con el linfangioma. El linfangioma se suele presentar en pacientes del mismo rango de edad y con una clínica de proptosis no inflamatoria y no dolorosa de igual evolución, y que es resultado de una hemorragia sobre una lesión existente que suele coincidir con un episodio de infección de vías respiratorias altas o un traumatismo orbitario previo. La característica diferencial es la presencia de otras lesiones linfangiomatosas extraorbitarias (como el paladar) y quistes hemorrágicos en la TC que no son típicos el RMS.
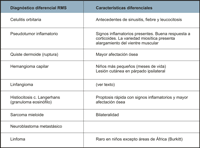
Tabla 1. Diagnóstico diferencial del rabdomiosarcoma. Características diferenciales.
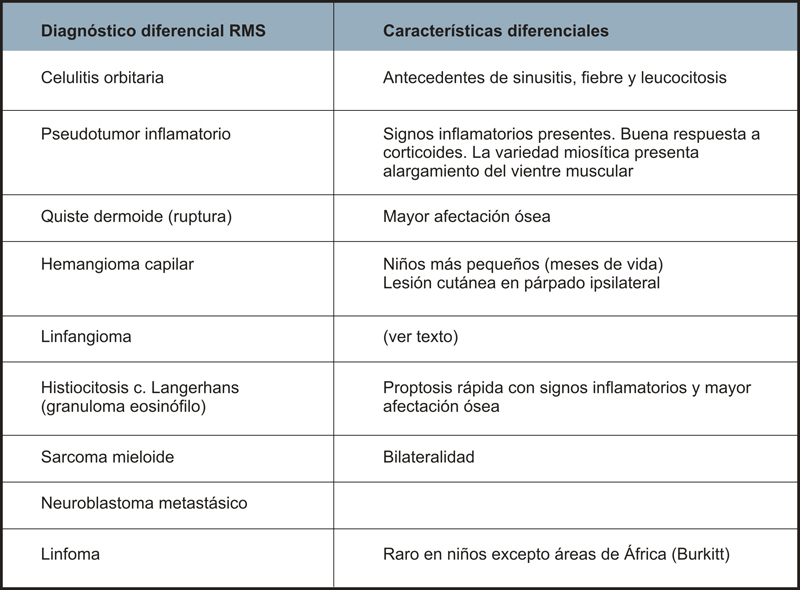
El rabdomiosarcoma orbitario puede extenderse a través de la órbita y la cavidad craneal tras una larga evolución de la lesión, pero actualmente las metástasis son raras, y si ocurren lo hacen vía hematógena preferentemente a pulmón y sistema óseo, afectando gravemente al pronóstico.
El tratamiento de elección hace poco más de 50 años consistía en la exanteración orbitaria y diversos regímenes quimioterápicos, con una tasa de mortalidad muy alta. Actualmente, y gracias a los ensayos clínicos randomizados realizados por el IRSG (Intergroup Rhabdomiosarcoma Study Group, (4292 RMS desde 1971) para el tratamiento del RMS, la supervivencia a los 5 años ha ido aumentando del 25% hasta llegar a un 70% (en 1991). El IRSG permite la clasificación de los rabdomiosarcomas en 4 estadios según su extensión al diagnóstico, y junto a la histología del tumor y, recientemente la inmunohistoquímica, permiten la selección de la terapia quimioterápica y radioterapia a realizar, así como la aproximación pronóstica. Se ha reconocido la localización orbitaria, como un factor pronóstico vital mejor que la aparición del RMS en otras localizaciones7.
Está en marcha el estudio IRS-V que combina la clasificación grupal, estadio, y subtipo histológico para clasificar los pacientes en 3 diferentes protocolos terapéuticos de acuerdo al riesgo de recurrencia.
Se debe realizar un seguimiento de los pacientes en tratamiento radioterápico para detectar las posibles complicaciones oftalmológicas como son: ojo seco, catarata por radiación, hipoplasia orbitaria, segundas neoplasias, retinopatía, neuropatía óptica. El cristalino es extremadamente sensible a la radiación y en un 90% se puede esperar el desarrollo de cataratas años después. El uso de dosis reducidas, protecciones y localizaciones precisas reduce la incidencia de estas complicaciones.
En el pronóstico de los pacientes con RMS influyen varios factores como la localización del tumor primario, la clasificación IRSG al diagnóstico (extensión tumoral), subtipo histopatológico, edad del paciente (menores de 1 año tienen peor pronóstico de causa desconocida), contenido DNA celular (ploidía), translocaciones específicas y respuesta terapéutica.
El grupo IRVH (Institut de Recerca de Vall d’Hebron de Barcelona) ha descubierto recientemente dos proteínas que junto a la vía Notch están implicadas en la metástasis del RMS: integrina α9 y N-cadherina, y que podrían ser futura diana terapéutica de nuevas terapias moleculares8.
Agradecimientos
Mariona Suñol (Departamento de Anatomía Patológica del Hospital Universitari Sant Joan de Déu).Bibliografía
- Shields JA, Shields CL. Rhabdomyosarcoma: Review for the Ophthalmologist. Surv Ophthalmol. 2003;48:39-57.
- Shields JA, Shields CL. et al. Clinical Spectrum of Primary Ophthalmic Rhabdomyosarcoma. Ophthalmology. 2001;108:2284-92.
- Backhouse OC, Cove P, Bowen DI: Rhabdomyosarcoma masked by orbital trauma. Int. J. Oral Maxillofac. Surg. 1997;26:374-5.
- Frayer WC, Enterline HT. Embryonal rhabdomyosarcoma of the orbit in children and young adults. Arch Ophthalmol. 1959;62:203.
- Staibano S, Franco R et al. Orbital Rhabdomyosarcoma: Relationship Between DNA Ploidy, P53, Bcl-2, MDR-1 and Ki67 (MIB1) Expression and Clinical Behavior Anticancer Research 2004;24:249-58.
- Hayes-Jordan A, Andrassy R. Rhabdomyosarcoma in children. Current Opinion in Pediatrics. 2009;21:373-8.
- Beverly Raney R, Maurer HM, et al. The Intergroup Rhabdomyosarcoma Study Group (IRSG): major lessons from the IRS-I through IRS-IV studies as background for the current IRS-V treatment protocols. Sarcoma. 2001;5:9-15.
- Masia A, Almazán-Moga A, et al. Notch-mediated induction of N-cadherin and a9-integrin confers higher invasive phenotype on rhabdomyosarcoma cells. British Journal of Cancer. 2012;107:1374-83.