Volumen 22 - Número 3 - Julio-Septiembre 2014
Mancha rojo-cereza en la población pediátrica: diagnóstico diferencial
G. Romeu Cerrillo, J. CatalÃ
Hospital Sant Joan de Déu, Esplugues de Llobregat. Barcelona
CORRESPONDENCIA
RESUMEN
Presentamos el caso de una niña de 14 meses de edad con retraso psicomotor y mioclonias de extremidades superiores desde nacimiento, remitida a oftalmología para estudio de fondo de ojo. En la exploración destaca una baja agudeza visual y la imagen "mancha rojo-cereza" muy evidente en ambos ojos.
La mancha rojo-cereza en niños orienta al oftalmólogo y al neurólogo hacia una enfermedad por depósito lisosomal de lípidos (lipoidosis) casi de forma patognomónica, descartando por ejemplo otro gran grupo de enfermedades por depósito como las mucopolisacaridosis (depósito de glicosaminoglicanos).
La mancha rojo-cereza en niños nos debe alertar de que estamos ante una enfermedad sistémica grave e incurable hasta el momento, con un pronóstico vital que muchas veces no supera los 5 años de edad.
La mancha rojo-cereza en niños orienta al oftalmólogo y al neurólogo hacia una enfermedad por depósito lisosomal de lípidos (lipoidosis) casi de forma patognomónica, descartando por ejemplo otro gran grupo de enfermedades por depósito como las mucopolisacaridosis (depósito de glicosaminoglicanos).
La mancha rojo-cereza en niños nos debe alertar de que estamos ante una enfermedad sistémica grave e incurable hasta el momento, con un pronóstico vital que muchas veces no supera los 5 años de edad.
RESUM
Presentem el cas d'una nena de 14 mesos d'edat amb retràs psicomotor i mioclonies d'extremitats superiors des de naixement, derivada a oftalmologia per estudi de fons d'ull. En l'exploració destaca una baixa agudesa visual i la imatge "taca vermell-cirera" molt evident en tots dos ulls.
La taca vermell-cirera en nens orienta a l'oftalmòleg i al neuròleg a una malaltia per dipòsit lisosomal de lípids (lipoidosi) casi de forma patognomònica, descartant per exemple un altre gran grup de malalties per dipòsit com les mucopolisacaridosis (dipòsit de glicosaminoglicans).
La taca vermell-cirera en nens ens ha d'alterar que estem davant una malaltia sistèmica greu i incurable fins a dia d'avui, amb un pronòstic vital que moltes vegades no supera els 5 anys d'edat.
La taca vermell-cirera en nens orienta a l'oftalmòleg i al neuròleg a una malaltia per dipòsit lisosomal de lípids (lipoidosi) casi de forma patognomònica, descartant per exemple un altre gran grup de malalties per dipòsit com les mucopolisacaridosis (dipòsit de glicosaminoglicans).
La taca vermell-cirera en nens ens ha d'alterar que estem davant una malaltia sistèmica greu i incurable fins a dia d'avui, amb un pronòstic vital que moltes vegades no supera els 5 anys d'edat.
ABSTRACT
We present a medical case of a girl of 14 months with a psychomotor delay and upper limbs myoclonia from birth. She was sent to Ophthalmology for a fundus eye exploration, where a low visual acuity was standing out and the image of a "cherry red spot” was noticeable in both eyes.
Kids' "cherry red spots” guides the ophthalmologist and neurologist to consider a lipid lysosomal storage disease (lipidosis), almost in a pathognomonic way, dismissing another group of storage diseases like mucopolysaccharidosis (glycosaminoglycans deposit)
Furthermore, kids "cherry red spots" must alert that we are finding a serious systemic and incurable disease for the moment with a vital prognosis that might not exceed 5 years old.
Kids' "cherry red spots” guides the ophthalmologist and neurologist to consider a lipid lysosomal storage disease (lipidosis), almost in a pathognomonic way, dismissing another group of storage diseases like mucopolysaccharidosis (glycosaminoglycans deposit)
Furthermore, kids "cherry red spots" must alert that we are finding a serious systemic and incurable disease for the moment with a vital prognosis that might not exceed 5 years old.
Caso clínico
Se trata de una niña de 14 meses de edad remitida para fondo de ojo desde neurología. La paciente está en estudio por retraso psicomotor y mioclonías de extremidades superiores, presentes desde nacimiento y que han ido aumentando en intensidad y frecuencia en los últimos meses. Los padres destacan sobresaltos exagerados y "cara de susto" ante ruidos, luces y estímulos táctiles.
En la exploración oftalmológica destaca una baja agudeza visual por su edad, unos potenciales evocados visuales desestructurados y un electroretinograma normal. En el fondo de ojo se objetiva la "mancha rojo-cereza" muy evidente en ambos ojos (Figura 1 y Figura 2).
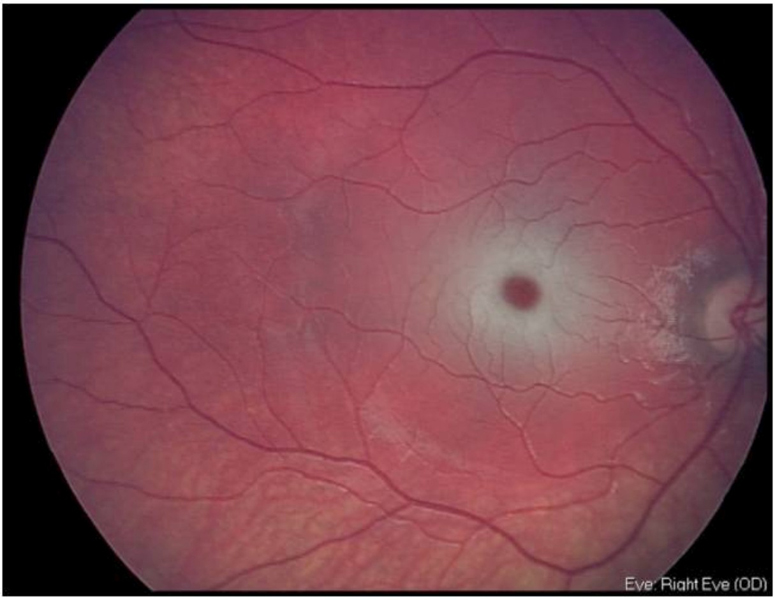
Figura 1. Mancha rojo-cereza ojo derecho.
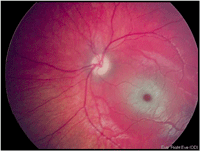
Figura 2. Mancha rojo-cereza ojo izquierdo.
El diagnóstico diferencial de la "mancha rojo-cereza" en la población pediátrica debe hacerse entre 3 grandes entidades: primero, la obstrucción de la arteria central de la retina (aunque muy infrecuente en niños y muy improbable si es bilateral); segundo, conmoción retiniana post-traumática que afecta al polo posterior (fácilmente descartable por anamnesis); y tercero, el gran grupo de enfermedades por depósito lisosomal (causa más frecuente en la infancia).
Gracias a la imagen funduscópica "mancha rojo-cereza" el diagnóstico se orientó rápidamente hacia una lipoidosis (enfermedad de Tay-Sachs en el caso de nuestra paciente), donde la disfunción de una enzima lisosomal conduce al depósito de lípidos en el sistema nervioso central, con la consecuente degeneración y muerte prematura del individuo afectado.
Discusión
La enfermedad de Tay-Sachs es una lipoidosis, es decir, existe un error innato en el metabolismo de los lípidos que provoca el acúmulo de esfingolípidos en los lisosomas de las células del SNC y de la médula espinal.Es una enfermedad muy poco frecuente (1/320.000), aunque 100 veces más prevalente entre los judíos de origen Ashkenazi (1/35.000).
Se debe a una mutación en el gen HEX-A (cromosoma 15), y se hereda de forma autosómica recesiva, siendo los heterocigotos portadores que no desarrollan la enfermedad.
Los pacientes con Tay-Sachs son incapaces de producir la enzima lisosómica HEXOSAMINIDASA-A (HEX-A), que en condiciones normales participa en la degradación de los gangliósidos GM2, componente lipídico de las membranas neuronales. El déficit de la HEX-A, pues, conlleva el acúmulo progresivo de gangliósidos en el SNC, su deterioro y la muerte de células nerviosas, y en última instancia también la muerte prematura del individuo afectado.
Manifestaciones clínicas
- La enfermedad de Tay-Sachs debuta con respuestas motoras anormales ante estímulos táctiles, luminosos y auditivos (mioclonias de extremidades y "cara de susto"). Aparece también de manera precoz hipotonia y retraso psicomotor. A los 12 meses de edad, el 100% de estos pacientes presentan la "mancha rojo-cereza" en el fondo de ojo, hasta el punto que su ausencia nos tiene que hacer dudar del diagnóstico. Más adelante se observa macrocefalia por depósito masivo de lípidos en el SNC.
- Existen 3 variantes clínicas, según el grado de disfunción de la enzima HEX-A (Tabla 1); la infantil es forma más frecuente con diferencia.

Tabla 1. Formas clínicas de Tay-Sachs según el grado de actividad de la HEX-A.

La enfermedad de Tay-Sachs se diagnostica midiendo la función enzimática de la HEX-A en suero, aunque la confirmación se hace con el análisis genético de la mutación (cromosoma 15). El fondo de ojo es imprescindible, pues la mancha rojo-cereza está presente en el 100% de los niños afectos mayores de 12 meses.
Hasta el momento es una enfermedad incurable, sin tratamiento y de muy mal pronóstico vital en las formas infantiles, pues conduce a la muerte prematura antes de los 5 años de edad.
Bibliografía recomendada
- Cruz M, Crespo M. Manual de pediatría. Ergon; 2003;Capitol 62: 335-343.
- Zasshi Y. Molecular pathogenesis and therapeutic approach of GM2 gangliosidosis. 2013;133(2):269-74.
- Bley AE, Giannikopoulos OA, Hayden D, Kubilus K, Tifft CJ, Eichler FS. Natural history of infantile G(M2) gangliosidosis. Pediatrics. 2011 Nov;128(5):e1233-41. doi: 10.1542/peds.2011-0078. Epub 2011 Oct 24.