Volumen 21 - Número 5 - Diciembre 2013
Tratamiento y seguimiento en el retinoblastoma
J. Català -Mora1, J. Abelairas2, G. Chantada3, J. DÃaz4, E. Escribano5, R. Matute6, N. MartÃn-Begué7, N. Pastora8, A. Parareda3, J. Peralta9 (Incluye vÃdeo)
1Responsable de la Unidad de Retinoblastoma. Unidad de Vítreo-Retina. Hospital Sant Joan de Déu. Esplugues de Llobregat. Barcelona. 2Profesor Asociado Universidad Autónoma de Madrid. Jefe de Sección Oftalmología infantil Hospital Universitario La Paz. Madrid. 3Hospital Sant Joan de Déu. Esplugues de Llobregat. Barcelona. 4Unidad Vítreo-Retina. Hospital Sant Joan de Déu. Esplugues de Llobregat. Barcelona. Hospital Sant Pau. Barcelona. 5Sección de Oncología Radioterápica infantil. Hospital Universitario La Paz. Madrid. 6Jefe de Sección de Oncología Radioterápica. Instituto Madrileño Oncología Clínica. La Milagrosa de Madrid. 7Unidad Oftalmología Pediátrica. Hospital Universitari Vall d’Hebron, Barcelona. 8Sección Retina Pediátrica. Hospital Infantil La Paz. Madrid. 9Oftalmología infantil Hospital Universitario La Paz. Madrid.
CORRESPONDENCIA
J. Català-Mora
E-mail: jcatalam@hsjdbcn.org
E-mail: jcatalam@hsjdbcn.org
El tratamiento del retinoblastoma es multidisciplinario e incluye la participación de diversos profesionales y de diferentes especialidades. No puede olvidarse que es una neoplasia potencialmente mortal. La prioridad absoluta es reducir la mortalidad de los pacientes, que en nuestro medio se centra en tres causas: metástasis, pinealoblastomas y segundos tumores. La quimioterapia tiene un papel fundamental en estas situaciones y ha disminuido el número de patologías tumorales en la pineal, desde la era posquimioterapia. En segundo lugar, el objetivo es mantener el globo ocular y finalmente conservar la visión.
La mayor parte de las exploraciones y de los tratamientos requieren anestesia general. En la actualidad, las modernas técnicas de anestesia inducida con intubación traqueal o mascarillas laríngeas son muy seguras, se realizan de manera ambulatoria y facilitan enormemente los múltiples tratamientos con la periodicidad diaria con que se utilizan. La sedación con gas Sevorane® es la que empleamos con nuestros pacientes.
En los tumores unilaterales muy grandes, en estadio E, con múltiples siembras vítreas o invasión de la cámara anterior, el tratamiento de elección es la enucleación. En los tumores unilaterales en estadios B y D se realiza quimiorreducción, ya sea sistémica o intraarterial según la experiencia y el protocolo de los diferentes centros, seguida de terapia de consolidación: crioterapia en los tumores anteriores pequeños (< 1,5 mm de grosor), termoterapia o quimiotermoterapia con láser de diodo en los tumores posteriores pequeños, y braquiterapia con rutenio o con yodo en los tumores intermedios y en casos de recidiva tras crioterapia o termoterapia.
En los tumores bilaterales el tratamiento inicial, excepto en ojos con estadio E muy avanzado que van directamente a enucleación, consiste en quimiorreducción mediante quimioterapia sistémica o intraarterial y posteriormente tratamiento de consolidación local, tal como antes se ha descrito.
Recientemente se ha incorporado la quimioterapia intraarterial, en la cual se inyecta melfalán, topotecán, carboplatino o una combinación directamente en la arteria oftálmica; es muy efectiva y con menores efectos secundarios sistémicos. Otras formas de administración de la quimioterapia, como subtenoniana, retroocular o intravítrea, se usan en casos seleccionados y sólo en centros de referencia.
Una vez realizada la quimioterapia debemos consolidar el tumor con las siguientes técnicas:
Es una técnica precisa, segura y fiable. Los tejidos sanos del niño en fase de desarrollo, como el osteoarticular y el orbitofacial, son muy sensibles. Con este método logramos disminuir los efectos secundarios y la dosis de radiación.
Otras opciones para el tratamiento con radioterapia externa son el acelerador de protones, el LINAC tridimensional y el Gamma Knife que se están desarrollando, aunque con poca experiencia en pediatría.
En la planificación previa al tratamiento se obtiene una TC sobre la cual se contornean el volumen diana (globo ocular) y los órganos de riesgo que hay que preservar de la irradiación lo máximo posible.
Cada día de tratamiento se obtienen imágenes de TC de megavoltaje (tomoimagen), que se fusionarán con las obtenidas en la fase de planificación y permitirán colocar al niño exactamente igual durante todas las sesiones.
El acelerador lineal, que gira 360º alrededor del paciente, irradia helicoidalmente con un número de segmentos entre 10.000 y 200.000, según el tamaño de la zona a tratar. Al dividir la radiación entre tantas puertas de entrada, es baja en los tejidos adyacentes que atraviesa y lo suficientemente intensa en la lesión, que es donde convergen.
Un caso típico es un paciente de 14 meses de edad al comenzar la tomoterapia, diagnosticado y enucleado de un ojo con 6 meses, y con retinoblastomas multifocales en el otro ojo, que precisó tratamiento quimioterápico, termoterapias y crioterapias múltiples. Se le colocó una placa de rutenio a los 9 meses y se realizaron nuevos tratamientos de consolidación. Fue derivado a radioterapia por aparición de reactivaciones y tumores periféricos. Bajo anestesia se inmoviliza al paciente con una mascarilla termoplástica individualizada, y se realiza una TC de planificación. Comienza el tratamiento de tomoterapia con fotones de 6 MEV, incluyendo el globo ocular, con una dosis total de 45 Gy, 180 cGy/fracción, una fracción al día y cinco días a la semana, con un total de 25 sesiones.
Debe considerarse la invasión del nervio óptico, de la órbita por contiguidad y del sistema nervioso central. Las metástasis hemáticas y linfáticas muestran, en las estadísticas, un pronóstico vital muy malo; sin embargo, se han publicado artículos con resultados sorprendentes combinando cirugías agresivas, quimioterapias y radioterapias en caso de afectación orbitaria (Figura 2).
Procedimientos intratecales, e incluso autotrasplante de médula ósea en casos ya muy desesperados, utilizan múltiples y nuevos fármacos con agentes inmunosupresores.
Si añadimos el factor genético a todos estos tratamientos, estamos favoreciendo la aparición de segundas neoplasias y tumores iatrogénicos.
Siempre debemos pensar en tumores de la línea media cerebral y retinoblastomas trilaterales (bilaterales + pinealoblastomas).
La exenteración orbitaria ha caído en desuso por la gran amputación anatómica y la disponibilidad de nuevas terapias menos deformantes.
La LIO nos artefactará la visión periferica por las opacidades en la cápsula, como puede verse en la imagen de Retcam (Figura 3).
El patrón de regresión parece que podría estar condicionado por el tratamiento empleado. Así, por ejemplo, cuando se utiliza radioterapia externa como tratamiento único, el patrón de regresión de tipo 1 es el más prevalente y el tamaño tumoral es el único factor estadísticamente significativo relacionado con el patrón de regresión. Cuando se utiliza quimiorreducción, el patrón de regresión viene condicionado por el tipo de tratamiento local de consolidación que se asocie, que a su vez viene determinado por el tamaño y la localización inicial de tumor. Los tumores de mayor tamaño y situados cerca de la fóvea suelen presentar un patrón de regresión de tipo 1 o 3, mientras que los tumores pequeños o localizados más periféricos presentan un patrón de regresión de tipo 4.
Los patrones de regresión de los tipos 2 y 3 son los más difíciles de diferenciar de un tumor activo. El seguimiento seriado permitirá hacer el diagnóstico diferencial: un tumor inactivo no crece y permanece sin cambios, mientras que uno activo crecerá de manera progresiva en 1 a 4 meses. De todos modos, los patrones de regresión pueden ir variando ligeramente con el tiempo, apreciándose pequeños cambios que no deben confundirse con actividad. El porcentaje de los patrones de regresión de los tipos 2 y 3 disminuyen con el tiempo y aumentan los tipos 0, 1 y 4.
El riesgo de una recidiva es independiente del tipo de patrón de regresión que haya presentado el tumor. Parece ser que el único factor predictivo de la recidiva del retinoblastoma es el tamaño tumoral y su localización. Las recidivas son más frecuentes durante el primer año, y no se han descrito tras 4 años de seguimiento en pacientes tratados con quimiorreducción asociada a tratamientos locales, ni en pacientes sometidos a radioterapia externa.
Cabe destacar que algunos retinoblastomas muestran mínimos cambios tras recibir tratamiento, lo que parece ser una característica de los tumores bien diferenciados. Entre estos tumores se incluyen los retinoblastomas que presentan cavidades en el espesor del tumor, las cuales se observan en la oftalmoscopia. Suelen disminuir poco su tamaño tras el tratamiento, con independencia del tratamiento empleado, y por otro lado no presentan muchas calcificaciones inicialmente ni tras el tratamiento. No hay que confundir su falta de respuesta al tratamiento con agresividad del tumor.
Es necesario indicar universalmente el diagnóstico molecular y el consejo genético, es decir, a todos los pacientes afectos de retinoblastoma. Aquellos con antecedentes familiares de retinoblastoma, con retinoblastoma bilateral y hasta un 15% de los afectos de retinoblastoma unilateral serán portadores de una mutación del gen RB.
La indicación de estudio genético a todos los pacientes de retinoblastoma permitirá conocer si ellos o sus familiares son portadores de una mutación y, por lo tanto, requieren controles clínicos por el riesgo de desarrollar más tumores, o bien, en caso de que no lo sean, pueden ahorrarse exámenes oftalmológicos y controles clínicos exhaustivos. La determinación también permitirá poder establecer un consejo genético documentado y, probablemente en un futuro, estratificar a los pacientes según el genotipo que pueda condicionar un mayor riesgo de desarrollo tumoral (Tabla 2, Tabla 3 y Tabla 4).
El tratamiento del retinoblastoma con quimioterapia involucra numerosas situaciones, desde la prevención del desarrollo de enfermedad extraocular en pacientes enucleados con factores histológicos de riesgo, hasta el tratamiento de la enfermedad metastásica, incluyendo asimismo un papel creciente en el tratamiento conservador de la visión, donde se utiliza por vía sistémica o localizada. Sin embargo, existen numerosas controversias en términos del valor pronóstico de los diferentes factores de riesgo histológico, así como sobre la composición y la intensidad de los regímenes a utilizar para la prevención de las metástasis. A la luz de los conocimientos actuales, se concluye que los pacientes enucleados inicialmente y que presentan invasión coroidea aislada tienen una sobrevida superior al 95%, incluyendo aquellos que presentan invasión masiva. En estos pacientes, el riesgo de recaída extraocular es significativamente mayor, aunque la sobrevida global no se ve afectada por la omisión de la quimioterapia adyuvante. Ésta es necesaria, sin embargo, cuando el tumor ha atravesado la coroides y ha penetrado en la esclera; de hecho, estos niños se benefician de un esquema de mayor intensidad de dosis. En los pacientes con invasión retrolaminar del nervio óptico, el uso de quimioterapia con esquemas de alta intensidad se asocia significativamente a una mejor sobrevida que el uso de regímenes menos intensos.
Un porcentaje de los pacientes que presentan enfermedad diseminada en la órbita pueden sobrevivir con tratamiento multimodal incluyendo quimioterapia y radioterapia convencionales asociadas a cirugía. El tratamiento de la enfermedad metastásica con dosis altas de quimioterapia seguida de rescate autólogo de progenitores hematopoyéticos ha dado resultados alentadores.
En cuanto al tratamiento conservador, hay mucha controversia acerca de cuál es el más efectivo y con menor morbilidad. La quimioterapia sistémica con regímenes con carboplatino y etopósido dan excelentes resultados en los ojos con enfermedad poco avanzada, aunque en aquellos con siembras vítreas los resultados no son favorables y suelen ser enucleados o irradiados. En los últimos años se han popularizado las vías locales de aplicación, con el fin de lograr una mayor concentración de quimioterapia en el ojo y en especial en el vítreo. La aplicación de quimioterapia por vía intraarterial en la arteria oftálmica ha resultado ser una terapia prometedora, logrando altas concentraciones de los fármacos y aumentando la tasa de preservación ocular en ojos con enfermedad avanzada. Sin embargo, al ser una técnica que requiere una alta concentración de recursos, es de difícil reproducibilidad y sólo se encuentra disponible en centros de alta complejidad. En los últimos años se ha visto un resurgimiento de la quimioterapia administrada directamente por inyección intravítrea, la cual durante años ha sido contraindicada por el riesgo de siembra orbitaria secundaria. En la actualidad, mediante el uso de una técnica modificada, parecería ser segura al administrar altas dosis de quimioterapia en el vítreo.
En los casos tratados con quimioterapia sistémica, en pacientes tanto con afectación congénita como adquirida, deberá controlarse también la función auditiva, por el uso de carboplatino, y la función medular, sobre todo cuando el tratamiento haya incluido etopósido.
Aparte de los efectos secundarios oculares, en los pacientes con retinoblastoma, sobre todo cuando se ha requerido una enucleación, está indicado un seguimiento neuropsicológico. El solo hecho de padecer un tumor ocular ya aumenta dicho riesgo.
Capítulo aparte en relación con el seguimiento merecen los pacientes con afectación congénita, es decir, aquellos que presenten predisposición por alteraciones del gen RB1. Como es bien sabido, estos pacientes tienen muchas posibilidades de que el retinoblastoma sea multifocal, y habitualmente bilateral. Este riesgo requiere un seguimiento frecuente por oftalmología, para descartar la aparición de nuevos tumores y poder tratarlos precozmente en caso de que aparezcan. El control debe ser estricto hasta los 5 años de edad, período en que el riesgo de aparición de tumores es mayor. En el seguimiento de estos pacientes deberá indicarse también una resonancia magnética cerebral semestral durante los 2 años siguientes al diagnóstico, y anual hasta los 5 años, para descartar un retinoblastoma trilateral, tumor neuroblástico de la línea media intracraneal, que aparece en un 5% a 15% de los pacientes con afectación congénita y que es una de las causas más frecuentes de mortalidad en nuestro medio.
Los pacientes con afectación congénita tienen también un mayor riesgo de padecer otros tumores a lo largo de su vida. Entre las segundas neoplasias más frecuentes se incluyen los osteosarcomas, fundamentalmente en las zonas irradiadas (por ello se aconseja no irradiar a estos pacientes antes de los 12 meses de edad), los sarcomas de partes blandas como el leiomiosarcoma (riesgo relativo de 390, con una mediana de aparición a los 30 años del diagnóstico), el fibrosarcoma, el rabdomiosarcoma y el liposarcoma, tumores cerebrales como los gliomas de alto grado, melanomas malignos, tumores epiteliales como los carcinomas de pulmón y de mama, y linfomas. El seguimiento de estos pacientes debería proponerse en unidades de seguimiento oncológico para poder descartar la presencia de segundas neoplasias ante cualquier clínica de sospecha (Figura 7 y Figura 8).
Con el aumento de las unidades de vitreorretina se están operando en nuestro país síndromes de enmascaramiento, hemorragias vítreas, endoftalmitis y casos extremos con desprendimiento de retina que resultan ser retinoblastomas. Al remitirnos a estos pacientes a los hospitales de referencia, se incrementa en nuestro medio la derivación para tomoterapia (Figura 9).
La mayor parte de las exploraciones y de los tratamientos requieren anestesia general. En la actualidad, las modernas técnicas de anestesia inducida con intubación traqueal o mascarillas laríngeas son muy seguras, se realizan de manera ambulatoria y facilitan enormemente los múltiples tratamientos con la periodicidad diaria con que se utilizan. La sedación con gas Sevorane® es la que empleamos con nuestros pacientes.
En los tumores unilaterales muy grandes, en estadio E, con múltiples siembras vítreas o invasión de la cámara anterior, el tratamiento de elección es la enucleación. En los tumores unilaterales en estadios B y D se realiza quimiorreducción, ya sea sistémica o intraarterial según la experiencia y el protocolo de los diferentes centros, seguida de terapia de consolidación: crioterapia en los tumores anteriores pequeños (< 1,5 mm de grosor), termoterapia o quimiotermoterapia con láser de diodo en los tumores posteriores pequeños, y braquiterapia con rutenio o con yodo en los tumores intermedios y en casos de recidiva tras crioterapia o termoterapia.
En los tumores bilaterales el tratamiento inicial, excepto en ojos con estadio E muy avanzado que van directamente a enucleación, consiste en quimiorreducción mediante quimioterapia sistémica o intraarterial y posteriormente tratamiento de consolidación local, tal como antes se ha descrito.
Quimioterapia
El retinoblastoma es un tumor quimiosensible. Habitualmente la quimioterapia se usa en tumores bilaterales y en casos unilaterales en los cuales no está indicada la enucleación de entrada. El objetivo es la quimiorreducción tumoral para poder aplicar tratamientos de consolidación local. Según el centro se administra de forma sistémica, habitualmente combinando dos o tres fármacos: carboplatino, vincristina y etopóxido, entre tres y ocho ciclos.Recientemente se ha incorporado la quimioterapia intraarterial, en la cual se inyecta melfalán, topotecán, carboplatino o una combinación directamente en la arteria oftálmica; es muy efectiva y con menores efectos secundarios sistémicos. Otras formas de administración de la quimioterapia, como subtenoniana, retroocular o intravítrea, se usan en casos seleccionados y sólo en centros de referencia.
Una vez realizada la quimioterapia debemos consolidar el tumor con las siguientes técnicas:
- Termoterapia transpupilar y quimiotermoterapia: mediante un láser diodo se aplica hipertermia sobre el tumor para provocar una apoptosis celular. Se indica en tumores postecuatoriales de tamaño pequeño y grosor menor de 2 mm. Si se combina con carboplatino (quimiotermoterapia) se consigue una mayor penetración (Video 1).
- Crioterapia: destrucción mediante triple criocongelación del tumor. Indicada en tumores anteriores pequeños con un grosor menor de 1,5 mm (Video 2).
- Braquiterapia: el retinoblastoma es un tumor muy radiosensible. Se utiliza preferentemente rutenio, aunque también pueden emplearse otros isótopos, como el yodo o el estroncio. Consiste en la aplicación de radiaciones ionizantes locales en la epiesclera para destruir el tumor. Se indica en tumores intermedios, de hasta 18 mm de diámetro y un grosor máximo de 4 mm (rutenio) o 6 mm (yodo) (Video 3).
- Radioterapia externa: consiste en la aplicación de radiaciones ionizantes sobre la órbita del paciente. Es una técnica muy efectiva, ya que el retinoblastoma es muy radiosensible. Entre sus efectos secundarios más frecuentes se encuentran la catarata, el síndrome seco, la atrofia orbitaria y los temidos segundos tumores en el área de radiación. Está contraindicada en los menores de 1 año y se reserva para aquellos casos bilaterales en que se necesite conservar la visión, o para recidivas, siembras vítreas o en caso de falta de respuesta con tratamientos de consolidación local.

Tabla 1. Seguimiento de los pacientes con retinoblastoma una vez completado el tratamiento y tras 1 año de estabilidad tumoral.

Radioterapia helicoidal en tiempo real guiada por la imagen (tomoterapia) para el tratamiento del retinoblastoma
La tomoterapia permite ajustar la radiación a la forma del globo ocular y a los tumores, combinando en un mismo equipo un sistema de obtención de imagen por tomografía computarizada (TC) y un acelerador lineal que gira helicoidalmente 360° alrededor del paciente. Así puede escalarse la dosis de radiación y minimizar la toxicidad sobre los órganos próximos.Es una técnica precisa, segura y fiable. Los tejidos sanos del niño en fase de desarrollo, como el osteoarticular y el orbitofacial, son muy sensibles. Con este método logramos disminuir los efectos secundarios y la dosis de radiación.
Otras opciones para el tratamiento con radioterapia externa son el acelerador de protones, el LINAC tridimensional y el Gamma Knife que se están desarrollando, aunque con poca experiencia en pediatría.
¿Cómo se realiza la tomoterapia? (Figura 1)

Figura 1.

En la planificación previa al tratamiento se obtiene una TC sobre la cual se contornean el volumen diana (globo ocular) y los órganos de riesgo que hay que preservar de la irradiación lo máximo posible.
Cada día de tratamiento se obtienen imágenes de TC de megavoltaje (tomoimagen), que se fusionarán con las obtenidas en la fase de planificación y permitirán colocar al niño exactamente igual durante todas las sesiones.
El acelerador lineal, que gira 360º alrededor del paciente, irradia helicoidalmente con un número de segmentos entre 10.000 y 200.000, según el tamaño de la zona a tratar. Al dividir la radiación entre tantas puertas de entrada, es baja en los tejidos adyacentes que atraviesa y lo suficientemente intensa en la lesión, que es donde convergen.
Planificación, dosimetría y fraccionamiento radioterápico de la tomoterapia
Los oncólogos radioterápicos eligen una dosis total de 40-45 Gy. Hay quien considera que podría variar entre 35 y 50 Gy. En casos avanzados y de gran tamaño se llega hasta 50-56 Gy. La dosis fracción varía entre 1,8 y 3,8 cGy durante 5 días a la semana.Un caso típico es un paciente de 14 meses de edad al comenzar la tomoterapia, diagnosticado y enucleado de un ojo con 6 meses, y con retinoblastomas multifocales en el otro ojo, que precisó tratamiento quimioterápico, termoterapias y crioterapias múltiples. Se le colocó una placa de rutenio a los 9 meses y se realizaron nuevos tratamientos de consolidación. Fue derivado a radioterapia por aparición de reactivaciones y tumores periféricos. Bajo anestesia se inmoviliza al paciente con una mascarilla termoplástica individualizada, y se realiza una TC de planificación. Comienza el tratamiento de tomoterapia con fotones de 6 MEV, incluyendo el globo ocular, con una dosis total de 45 Gy, 180 cGy/fracción, una fracción al día y cinco días a la semana, con un total de 25 sesiones.
Complicaciones
Como complicaciones se han observado conjuntivitis y dermatitis. No apreciamos por el momento en nuestra casuística, después de 2 años, retinopatía radiactiva, ojo seco, pérdida de pestañas ni catarata. En el desarrollo orbitofacial no observamos hasta hoy hipoplasia ósea. Ha aparecido alguna recidiva y hemos visto casos resistentes a la radioterapia que tuvieron que ser enucleados.La órbita, el retinoblastoma y el mas allá
Aunque por suerte en el momento actual se ven muy pocas extensiones del retinoblastoma fuera de las cubiertas oculares, hay que tenerlas presentes. En estas circunstancias, la tomoterapia como complemento puede ser necesaria y efectiva.Debe considerarse la invasión del nervio óptico, de la órbita por contiguidad y del sistema nervioso central. Las metástasis hemáticas y linfáticas muestran, en las estadísticas, un pronóstico vital muy malo; sin embargo, se han publicado artículos con resultados sorprendentes combinando cirugías agresivas, quimioterapias y radioterapias en caso de afectación orbitaria (Figura 2).

Figura 2. Imágenes de resonancia magnética de diferentes casos de retinoblastoma con extensión extraocular.

Procedimientos intratecales, e incluso autotrasplante de médula ósea en casos ya muy desesperados, utilizan múltiples y nuevos fármacos con agentes inmunosupresores.
Si añadimos el factor genético a todos estos tratamientos, estamos favoreciendo la aparición de segundas neoplasias y tumores iatrogénicos.
Siempre debemos pensar en tumores de la línea media cerebral y retinoblastomas trilaterales (bilaterales + pinealoblastomas).
La exenteración orbitaria ha caído en desuso por la gran amputación anatómica y la disponibilidad de nuevas terapias menos deformantes.
¿Qué sucede con las cataratas en el retinoblastoma? LIO sí, LIO no
Sigue habiendo una gran controversia en la literatura. Hay autores que operan con capsulorrexis amplias y vitrectomía anterior, analizando lo aspirado por citología y anatomía patológica; en otros centros se dilata la intervención en la medida de lo posible hasta la estabilización tumoral y se realiza facoaspiración, con implante de una lente intraocular (LIO) en niños mayores de 1 año sin capsulotomía posterior. Tras la radioterapia externa la opacificación capsular es rara y poco importante. En caso necesario, en niños mayores puede realizarse la capsulotomía con YAG.La LIO nos artefactará la visión periferica por las opacidades en la cápsula, como puede verse en la imagen de Retcam (Figura 3).

Figura 3. Múltiples artefactos en la valoración de fondo de ojo de un paciente operado de catarata e implante de lente intraocular tras radioterapia por retinoblastoma.
Patrones de regresión del retinoblastoma
Cuando se decide tratar un retinoblastoma con cualquier opción terapéutica que no sea la enucleación es importante conocer cuáles son los patrones de curación que puede presentar el tumor. Es raro que desaparezca completamente; por el contrario, lo más habitual es que persista una masa residual. Se han descrito cinco patrones de regresión:- Tipo 0: desaparición completa del tumor.
- Tipo 1: tumor totalmente calcificado (Figura 4).
- Tipo 2: tumor de aspecto translúcido sin ninguna calcificación.
- Tipo 3: tumor parcialmente calcificado, que comparte características de los patrones de regresión 1 y 2 (Figura 5).
- Tipo 4: cicatriz coriorretiniana plana (Figura 6).

Figura 4. Patrón de regresión de tipo 1. Se observa un resto tumoral totalmente calcificado en el polo posterior y siembras vítreas también calcificadas inferiores. También puede apreciarse la importante alteración del epitelio retiniano en todo el polo posterior y alrededor de las arcadas vasculares temporales.


Figura 5. Patrón de regresión de tipo 3. Se aprecia un resto tumoral inferior a la papila de aspecto translúcido con alguna calcificación intratumoral.


Figura 6. Patrón de regresión de tipo 4. Se aprecia una cicatriz coriorretinana en el área macular en un retinoblastoma tratado con varias sesiones de termoterapia transpupilar y radioterapia externa.

El patrón de regresión parece que podría estar condicionado por el tratamiento empleado. Así, por ejemplo, cuando se utiliza radioterapia externa como tratamiento único, el patrón de regresión de tipo 1 es el más prevalente y el tamaño tumoral es el único factor estadísticamente significativo relacionado con el patrón de regresión. Cuando se utiliza quimiorreducción, el patrón de regresión viene condicionado por el tipo de tratamiento local de consolidación que se asocie, que a su vez viene determinado por el tamaño y la localización inicial de tumor. Los tumores de mayor tamaño y situados cerca de la fóvea suelen presentar un patrón de regresión de tipo 1 o 3, mientras que los tumores pequeños o localizados más periféricos presentan un patrón de regresión de tipo 4.
Los patrones de regresión de los tipos 2 y 3 son los más difíciles de diferenciar de un tumor activo. El seguimiento seriado permitirá hacer el diagnóstico diferencial: un tumor inactivo no crece y permanece sin cambios, mientras que uno activo crecerá de manera progresiva en 1 a 4 meses. De todos modos, los patrones de regresión pueden ir variando ligeramente con el tiempo, apreciándose pequeños cambios que no deben confundirse con actividad. El porcentaje de los patrones de regresión de los tipos 2 y 3 disminuyen con el tiempo y aumentan los tipos 0, 1 y 4.
El riesgo de una recidiva es independiente del tipo de patrón de regresión que haya presentado el tumor. Parece ser que el único factor predictivo de la recidiva del retinoblastoma es el tamaño tumoral y su localización. Las recidivas son más frecuentes durante el primer año, y no se han descrito tras 4 años de seguimiento en pacientes tratados con quimiorreducción asociada a tratamientos locales, ni en pacientes sometidos a radioterapia externa.
Cabe destacar que algunos retinoblastomas muestran mínimos cambios tras recibir tratamiento, lo que parece ser una característica de los tumores bien diferenciados. Entre estos tumores se incluyen los retinoblastomas que presentan cavidades en el espesor del tumor, las cuales se observan en la oftalmoscopia. Suelen disminuir poco su tamaño tras el tratamiento, con independencia del tratamiento empleado, y por otro lado no presentan muchas calcificaciones inicialmente ni tras el tratamiento. No hay que confundir su falta de respuesta al tratamiento con agresividad del tumor.
Aspectos oncológicos en el tratamiento del retinoblastoma: estudio genético, quimioterapia y seguimiento a corto y largo plazo
Estudio genético de los pacientes con retinoblastoma
El retinoblastoma es un paradigma de predisposición genética al cáncer. Aproximadamente un 60% de los casos no son hereditarios y por lo tanto corresponden a tumores unifocales y unilaterales, y los pacientes no son transmisores de la mutación a su descendencia, ya que la mutación sólo estaría presente en las células tumorales. Del 40% restante de los casos, hereditarios, en torno a un 70% serán bilaterales y un 30% unilaterales. El 90% de los pacientes que hereden una mutación del gen RB sufrirán un retinoblastoma, casi siempre bilateral. De todos los retinoblastomas hereditarios, sólo una cuarta parte lo será a consecuencia de la transmisión de la mutación por parte de uno de los progenitores, y las tres cuartas partes restantes serán mutaciones que aparecerán de novo.Es necesario indicar universalmente el diagnóstico molecular y el consejo genético, es decir, a todos los pacientes afectos de retinoblastoma. Aquellos con antecedentes familiares de retinoblastoma, con retinoblastoma bilateral y hasta un 15% de los afectos de retinoblastoma unilateral serán portadores de una mutación del gen RB.
La indicación de estudio genético a todos los pacientes de retinoblastoma permitirá conocer si ellos o sus familiares son portadores de una mutación y, por lo tanto, requieren controles clínicos por el riesgo de desarrollar más tumores, o bien, en caso de que no lo sean, pueden ahorrarse exámenes oftalmológicos y controles clínicos exhaustivos. La determinación también permitirá poder establecer un consejo genético documentado y, probablemente en un futuro, estratificar a los pacientes según el genotipo que pueda condicionar un mayor riesgo de desarrollo tumoral (Tabla 2, Tabla 3 y Tabla 4).

Tabla 2. Riesgo de la futura descendencia de desarrollar un retinoblastoma en caso de historial familiar negativo.


Tabla 3. Riesgo de la futura descendencia de desarrollar un retinoblastoma en caso de historial familiar positivo.


Tabla 4. Riesgo de desarrollar tumor por parte de los familiares de un paciente con retinoblastoma.

Tratamiento oncológico del retinoblastoma
El tratamiento del retinoblastoma contempla dos situaciones principales: en primer lugar, como en toda enfermedad maligna, debe administrarse un tratamiento que salve la vida del paciente, lo cual en ocasiones incluye la enucleación del ojo afectado o el tratamiento multidisciplinario del paciente con enfermedad diseminada extraocular, que es más frecuente en los países en desarrollo y es la principal causa de muerte en ese contexto. El segundo escenario incluye el tratamiento conservador del ojo, cuando es posible, lo cual constituye un desafío ya que no sólo debe considerarse la eficacia del tratamiento sino también la posibilidad que estos tratamientos causen secuelas a largo plazo, como la aparición de segundos tumores.El tratamiento del retinoblastoma con quimioterapia involucra numerosas situaciones, desde la prevención del desarrollo de enfermedad extraocular en pacientes enucleados con factores histológicos de riesgo, hasta el tratamiento de la enfermedad metastásica, incluyendo asimismo un papel creciente en el tratamiento conservador de la visión, donde se utiliza por vía sistémica o localizada. Sin embargo, existen numerosas controversias en términos del valor pronóstico de los diferentes factores de riesgo histológico, así como sobre la composición y la intensidad de los regímenes a utilizar para la prevención de las metástasis. A la luz de los conocimientos actuales, se concluye que los pacientes enucleados inicialmente y que presentan invasión coroidea aislada tienen una sobrevida superior al 95%, incluyendo aquellos que presentan invasión masiva. En estos pacientes, el riesgo de recaída extraocular es significativamente mayor, aunque la sobrevida global no se ve afectada por la omisión de la quimioterapia adyuvante. Ésta es necesaria, sin embargo, cuando el tumor ha atravesado la coroides y ha penetrado en la esclera; de hecho, estos niños se benefician de un esquema de mayor intensidad de dosis. En los pacientes con invasión retrolaminar del nervio óptico, el uso de quimioterapia con esquemas de alta intensidad se asocia significativamente a una mejor sobrevida que el uso de regímenes menos intensos.
Un porcentaje de los pacientes que presentan enfermedad diseminada en la órbita pueden sobrevivir con tratamiento multimodal incluyendo quimioterapia y radioterapia convencionales asociadas a cirugía. El tratamiento de la enfermedad metastásica con dosis altas de quimioterapia seguida de rescate autólogo de progenitores hematopoyéticos ha dado resultados alentadores.
En cuanto al tratamiento conservador, hay mucha controversia acerca de cuál es el más efectivo y con menor morbilidad. La quimioterapia sistémica con regímenes con carboplatino y etopósido dan excelentes resultados en los ojos con enfermedad poco avanzada, aunque en aquellos con siembras vítreas los resultados no son favorables y suelen ser enucleados o irradiados. En los últimos años se han popularizado las vías locales de aplicación, con el fin de lograr una mayor concentración de quimioterapia en el ojo y en especial en el vítreo. La aplicación de quimioterapia por vía intraarterial en la arteria oftálmica ha resultado ser una terapia prometedora, logrando altas concentraciones de los fármacos y aumentando la tasa de preservación ocular en ojos con enfermedad avanzada. Sin embargo, al ser una técnica que requiere una alta concentración de recursos, es de difícil reproducibilidad y sólo se encuentra disponible en centros de alta complejidad. En los últimos años se ha visto un resurgimiento de la quimioterapia administrada directamente por inyección intravítrea, la cual durante años ha sido contraindicada por el riesgo de siembra orbitaria secundaria. En la actualidad, mediante el uso de una técnica modificada, parecería ser segura al administrar altas dosis de quimioterapia en el vítreo.
Seguimiento del paciente con antecedentes de retinoblastoma
El seguimiento del paciente con antecedentes de retinoblastoma debe ser diferencial en función de si la afectación es congénita o adquirida. En los casos de afectación adquirida, el seguimiento debe hacer hincapié fundamentalmente en las alteraciones oculares. Los riesgos ocular y visual, y por lo tanto su seguimiento, son muy importantes en los pacientes que han tenido un retinoblastoma. El tratamiento con radioterapia ocular u orbitaria aumenta el riesgo de efectos secundarios oculares, entre los que se encuentran xeroftalmía, atrofia del conducto lagrimal, retinopatía y maculopatía, queratitis, telangiectasias e hipoplasia orbitaria. Las cataratas serían también uno de los efectos secundarios con riesgo visual más frecuentes en los pacientes de retinoblastoma irradiados. Todos los pacientes enucleados requieren igualmente un seguimiento oftalmológico especializado.En los casos tratados con quimioterapia sistémica, en pacientes tanto con afectación congénita como adquirida, deberá controlarse también la función auditiva, por el uso de carboplatino, y la función medular, sobre todo cuando el tratamiento haya incluido etopósido.
Aparte de los efectos secundarios oculares, en los pacientes con retinoblastoma, sobre todo cuando se ha requerido una enucleación, está indicado un seguimiento neuropsicológico. El solo hecho de padecer un tumor ocular ya aumenta dicho riesgo.
Capítulo aparte en relación con el seguimiento merecen los pacientes con afectación congénita, es decir, aquellos que presenten predisposición por alteraciones del gen RB1. Como es bien sabido, estos pacientes tienen muchas posibilidades de que el retinoblastoma sea multifocal, y habitualmente bilateral. Este riesgo requiere un seguimiento frecuente por oftalmología, para descartar la aparición de nuevos tumores y poder tratarlos precozmente en caso de que aparezcan. El control debe ser estricto hasta los 5 años de edad, período en que el riesgo de aparición de tumores es mayor. En el seguimiento de estos pacientes deberá indicarse también una resonancia magnética cerebral semestral durante los 2 años siguientes al diagnóstico, y anual hasta los 5 años, para descartar un retinoblastoma trilateral, tumor neuroblástico de la línea media intracraneal, que aparece en un 5% a 15% de los pacientes con afectación congénita y que es una de las causas más frecuentes de mortalidad en nuestro medio.
Los pacientes con afectación congénita tienen también un mayor riesgo de padecer otros tumores a lo largo de su vida. Entre las segundas neoplasias más frecuentes se incluyen los osteosarcomas, fundamentalmente en las zonas irradiadas (por ello se aconseja no irradiar a estos pacientes antes de los 12 meses de edad), los sarcomas de partes blandas como el leiomiosarcoma (riesgo relativo de 390, con una mediana de aparición a los 30 años del diagnóstico), el fibrosarcoma, el rabdomiosarcoma y el liposarcoma, tumores cerebrales como los gliomas de alto grado, melanomas malignos, tumores epiteliales como los carcinomas de pulmón y de mama, y linfomas. El seguimiento de estos pacientes debería proponerse en unidades de seguimiento oncológico para poder descartar la presencia de segundas neoplasias ante cualquier clínica de sospecha (Figura 7 y Figura 8).

Figura 7


Figura 8.

Retos actuales
El retinoblastoma es y será por muchos años objeto de controversia en cuanto a sus múltiples tratamientos y clasificaciones. Conseguir que un niño recién nacido con un retinoblastoma bilateral y multifocal alcance el año de edad, cuando sabemos que la radioterapia va a ser la última solución, es uno de los mayores retos con que nos enfrentamos actualmente.Con el aumento de las unidades de vitreorretina se están operando en nuestro país síndromes de enmascaramiento, hemorragias vítreas, endoftalmitis y casos extremos con desprendimiento de retina que resultan ser retinoblastomas. Al remitirnos a estos pacientes a los hospitales de referencia, se incrementa en nuestro medio la derivación para tomoterapia (Figura 9).

Figura 9. El diagnóstico tardío y las vitrectomías en ojos de pacientes con retinoblastoma ensombrecen el pronóstico de estos casos.
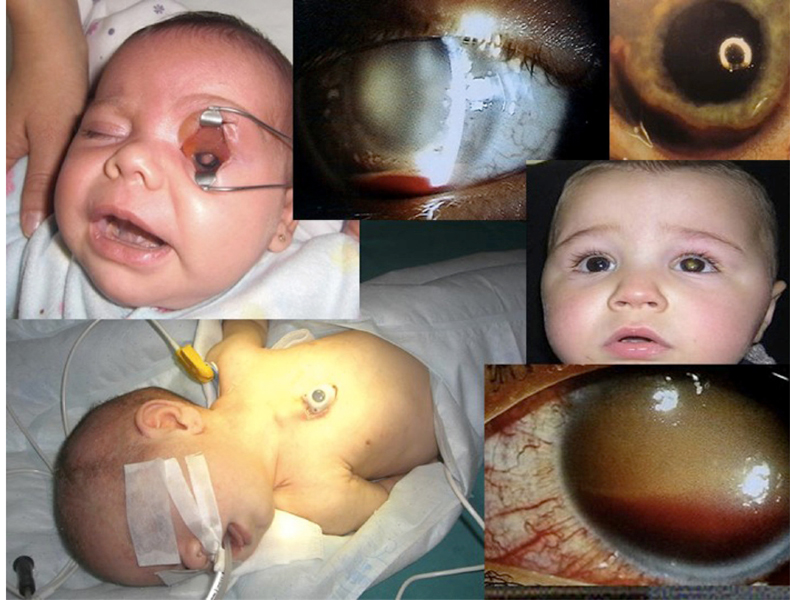
¿Es ético intentar salvar un globo ocular por todos los medios posibles?
Con la globalización, chats, blogs e Internet, las familias se ofuscan, quieren que se conserve el ojo a sus hijos y en algunos casos nos obligan a utilizar todos los tratamientos conservadores. Hay que suministrar consentimientos informados y hablar de las complicaciones añadidas. Los padres de los pacientes se relacionan e intercambian sus experiencias con angustia. Recomendar una enucleación a tiempo evita complicaciones innecesarias. Pese a todo, la espada de Damocles está sobre nuestras cabezas y estamos seguros de que en un futuro próximo nos veremos envueltos en litigios de juzgados, porque cada vez estamos menos protegidos por nuestros dirigentes hospitalarios y hay familiares de niños oncológicos que desarrollan una agresividad extrema.Consideraciones
- Pueden aparecer tumores en el ojo contralateral, incluso a los 5 años de haber enucleado un ojo.
- La aparición de retinoblastomas en la edad adulta y en nuestra experiencia con 7-8 años de edad nos hace ser muy cautos.
- Los tumores bilaterales y multifocales en los niños pequeños pueden evolucionar arbitrariamente y no podemos predecirlo.
- Aparecen nuevos tumores mientras los iniciales responden al tratamiento.
- Distintos retinoblastomas en el mismo ojo tienen diferentes respuestas en un mismo tiempo.
- Hay una alta recurrencia de focos vítreos y siembras subretinianas con la quimioterapia. Por suerte, con las nuevas técnicas de radiación, éstas son menores.
- Las terapias de consolidación focal no solucionan todos los casos.
Bibliografía recomendada
- Hosten N, Bornfeld N. Imaging of the globe and orbit; a guide to differential diagnosis. Stuttgart: Thieme; 1998. p.182-91.
- Català-Mora J, Parareda-Salles A, et al. Síndrome mascarada por retinoblastoma difuso. Arch Soc Esp Oftalmol. 2009;84:477-80.
- Singh AD, Shields CL, Shields JA. Prognostic factors in retinoblastoma. J Pediatr Ophthalmol Strabismus. 2000;37:134-41.
- Finger PT, Harbour JW, Karcioglu ZA. Risk factors for metastasis in retinoblastoma. Surv Ophthalmol. 2002;47:1-16.
- Dunphy EB. The story of retinoblastoma. Am J Ophthalmol. 1964;58:539-52.
- Singh AD, Garway-Heath D, et al. Relationship of regression pattern to recurrence in retinoblastoma. Br J Ophthalmol. 1993;77:12-6.
- Shields CL, Palamar M, et al. Retinoblastoma regression patterns following chemoreduccion and adjuvant therapy in 557 tumors. Arch Ophthalmol. 2009;127:282-90.
- Abramson DH, Gerardi CM, et al. Radiation regression patterns in treated retinoblastoma: 7 to 21 years later. J Pediatr Ophthalmol Strabismus. 1991;28:108-12.
- Shields CL, Mashayekhi A, et al. Chemoreduction for retinoblastoma. Analysis of tumor control and risks for recurrence in 457 tumors. Am J Ophthalmol. 2004;138:329-37.
- Mashayekhi A, Shields CL, Eagle RC, Shields JA. Cavitary changes in retinoblastoma: relationship to chemoresistance. Ophthalmology. 2005;112:1145-50.
- Rojanaporn D, Kaliki S, et al. Intravenous chemoreduction or intra-arterial chemotherapy for cavitary retinoblastoma: long-term results. Arch Ophthalmol. 2012;130:585-90.
- Atchaneeyasakul L, Murphree AL. Retinoblastoma. En: Ryan SJ, ed. Retina. 3rd ed. St Louis: Mosby; 2001. p. 513.
- Font RL, Croxatto JO, Rao NA. Tumors of the Eye and Ocular Adnexa, Atlas of Tumor Pathology. 4rd series, Fascicle 5. Washington, DC: AFIP; 2006. p. 86-103.
- Abdel Razek AA, Elkhamary S, Al-Mesfer S, Alkatan HM. Correlation of apparent diffusion coefficient at 3T with prognostic parameters of retinoblastoma. AJNR. 2012;33(5):944-8.
- Razek A, Elkhamary S, Mousa A. Differentiation between benign and malignant orbital tumors at 3-T diffusion MR-imaging. Neuroradiology. 2011;53(7):517-22.
- Elkhamary S, Song K, et al. Can preoperative MR imaging predict optic nerve invasion of retinoblastoma? Eur J Radiol. 2012; http://dx.doi.org/10.1016/j.ejrad.2012.03.034.
- Ventura G, Cozzi G. Red reflex examination for retinoblastoma. Lancet. 2012; 380(9844):803.
- De Graaf P, Pouwels PJW, Rodjan F, et al. Single-shot turbo spin-echo diffusion weighted imaging for retinoblastoma: initial experience. AJNR. 2012;33(1):110-8.
- Abelairas-Gómez J, Peralta-Calvo J, Sánchez-Jacob E, Gayá-Moreno F. Introducción al retinoblastoma. Actualización en Cirugía Oftálmica Pediátrica. Capítulo 23. LXXVI Ponencia Oficial de la Sociedad Española de Oftalmología. 2000;23:265-76.
- De Graaf P, van der Valk P, Moll A, Imhof S, Schoutenvan A, Meeteren V, et al. Contrast-enhancement of the anterior eye segment in patients with retinoblastoma: correlation between clinical, MR imaging, and histopathologic findings. AJNR. 2010;31:237-45.
- Saket RR, Mafee MF. Anterior-segment retinoblastoma mimicking pseudoinflammatory angle-closure glaucoma: review of the literature and the important role of imaging. AJNR. 2009;30:1607-9.
- Gorospe L, Royo A, Berrocal T, García-Raya P, Moreno P, Abelairas J. Imaging of orbital disorders in pediatric patients. Eur Radiol. 2003;13:2012-6.
- Chua J, Wisam J, Muen J, Reddy A, Brookes J. The masquerades of a childhood ciliary body medulloepithelioma: a case of chronic uveitis, cataract, and secondary glaucoma. Case Rep Ophthalmol Med. 2012;2012:493493.
- Wilson MW, Rodriguez-Galindo C, Billups C, et al. Lack of correlation between the histologic and magnetic resonance imaging results of optic nerve involvement in eyes primarily enucleated for retinoblastoma. Ophthalmology. 2009;116:1558-63.
- Moll AC, Hoekstra OS, Imhof SM, et al. Fluorine-18 fluorodeoxyglucose positron emission tomography (PET) to detect vital retinoblastoma in the eye: preliminary experience. Ophthalmic Genet. 2004;25:31-5.
- Dunkel IJ, Khakoo Y, Kernan NA, et al. Intensive multimodality therapy for patients with stage 4a metastatic retinoblastoma. Pediatr Blood Cancer. 2010;55:55-9.
- Brisse H, Guesmi M, Aerts I, Sastre-Garau X, Savignoni A, Lumbrosole Rouic L, et al. Relevance of CT and MRI in retinoblastoma for the diagnosis of postlaminar invasion with normal-size optic nerve: a retrospective study of 150 patients with histologic comparison. Pediatr Radiol. 2007;37:649-56.
- Silverman RH. High-resolution ultrasound imaging of the eye - a review. Clin Exp Ophthalmol. 2009;37:54-7.
- Song KD, Eo H, Kim JH, Yoo SY, Jeon TY. Can preoperative MR imaging predict optic nerve invasion of retinoblastoma? Eur J Radiol. 2012;81(12):4041-5.
- Finger PT, Meskin SW, Wisnicki HJ, et al. High frequency ultrasound of anterior segment retinoblastoma. Am J Ophthalmol. 2004;137:944-6.
- Shields CL, Mashayekhi A, Luo CK, et al. Optical coherence tomography in children: analysis of 44 eyes with intraocular tumors and simulating conditions. J Pediatr Ophthalmol Strabismus. 2004;41(6):338-44.
- Ruggeri M, Tsechpenakis G, Jiao S, et al. Retinal tumor imaging and volume quantification in mouse model using spectral-domain optical coherence tomography. Opt Express. 2009;17(5):4074-83.
- Dhoot D, Weissman J, Landon R, Evans M, Stout T. Optic nerve enhancement in Coats disease with secondary glaucoma. JAAPOS. 2009;13:301-2.
- Galluzzi P, Hadjistilianou T, Cerase A, De Francesco S, Toti P, Venturi C. Is CT still useful in the study protocol of retinoblastoma? AJNR. 2009;30(9):1760-5.
- Lemke A, Kazi I, Mergner U, Foerster P, Heimann H, Bechrakis N, et al. Retinoblastoma — MR appearance using a surface coil in comparison with histopathological results. Eur Radiol. 2007;17:49-60.
- De Graaf P, Knol D, Moll A, Imhof S, Schouten-van Meeteren A, Castelijns J. Eye size in retinoblastoma: MR imaging measurements in normal and affected eyes. Radiology. 2007;244:273-80.
- Lope L, Hutcheson K, Khademian Z. Magnetic resonance imaging in the analysis of pediatric orbital tumors: utility of diffusion-weighted imaging. JAAPOS. 2010;14(3):257-62.
- Abramson D, Beaverson C, Sangani P, et al. Screening for retinoblastoma: presenting signs as prognosticators of patient and ocular survival. Pediatrics. 2003;112:1248-55.
- Chantada G, Qaddoumi I, Canturk S, et al. Strategies to manage retinoblastoma in developing countries. Pediatr Blood Cancer. 2011;56:341-8.
- Radhakrishnan V, Kumar R, Arun Malhotra A, Bakhshil S. Role of PET/CT in staging and evaluation of treatment response after 3 cycles of chemotherapy in locally advanced retinoblastoma: a prospective study. J Nucl Med. 2012;53:191-8.
- Gatta G, Capoaccia R, Stiller C, et al. Childhood cancer survival trends in Europe: a EUROCARE Working Group Study. J Clin Oncol. 2005;23:3742-51.
- Shields CL, Honavar S, Shields JA, Demirci H. Vitrectomy in eyes with unsuspected retinoblastoma. Ophthalmology. 2000;107:2250-5.
- Palma J, Sasso DF, Dufort G, Koop K, Sampor C, Diez B, et al. Bone successful treatment of metastatic retinoblastoma with high-dose chemotherapy and autologous stem cell rescue in South America. Bone Marrow Transplant. 2012;47(4):522-7.
- Shields JA, Shields CL. Management of retinoblastoma. En: Intraocular tumors: an atlas and textbook. 2nd ed. Philadelphia: Lippincott Williams and Wilkins; 2008. p. 334-51.
- Atalar B, Ozyar E, Kaan Gunduz K, Gungor G. Intensity modulated radiotherapy (IMRT) in bilateral retinoblastoma. Radiol Oncol. 2010;44(3):194-8.
- McCormick B, Ellsworth R, Abramson D, LoSasso T, Grabowski E. Results of external beam radiation for children with retinoblastoma: a comparison of two techniques. J Pediatr Ophthalmol Strabismus. 1989;26:239-43.
- Schipper J. An accurate and simple method for megavoltage irradiation therapy of retinoblastoma. Radiother Oncol. 1983;1:31-41.
- MacCarthy A, Birch JM, Draper GJ, Hungerford JL, Kingston JE, Kroll ME, et al. Retinoblastoma in Great Britain 1963-2002. Br J Ophthalmol. 2009;93:33-7.
- Mallipatna AC, Sutherland JE, Gallie BL, Chan H, Héon E. Management and outcome of unilateral retinoblastoma. JAAPOS. 2009;13(6):546-50.
- Kivelä T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. Br J Ophthalmol. 2009;93:1129-31.
- Abramson DH, Du TT, Beaverson KL. (Neonatal) retinoblastoma in the first month of life. Arch Ophthalmol. 2002;120(6):738-42.
- Krengli M, Liebsch NJ, Hug EB, Orecchia R. Review of current protocols for proton therapy in USA. Tumori. 1998;84(2):209-16.
- Shields CL, Ramasubramanian A, Thangappan A, Hartzell K, Leahey A, Meadows AT, et al. Chemoreduction for group E retinoblastoma. Comparison of chemoreduction alone versus chemoreduction plus low dose external radiotherapy in 76 eyes. Ophthalmology. 2009;116:544-51.
- Dimaras H, Kimani K, Dimba EA, et al. Retinoblastoma. Lancet. 2012;379(9824):1436-46.
- Mehta M, Sethi S, Pushker N, Kashyap S, Sen S, Bajaj MS, et al. Retinoblastoma. Singapore Med J. 2012;53(2):128-36.
- Grabowska A, Calvo JP, Fernández-Zubillaga A, Ríos JC, Gómez JA. A magnetic resonance imaging diagnostic dilemma: diffuse infiltrating retinoblastoma versus Coats’ disease. J Pediatr Ophthalmol Strabismus. 2010;47.
- Shields CL, Ghassemi F, Tuncer S, Thangappan A, Shields JA. Clinical spectrum of diffuse infiltrating retinoblastoma in 34 consecutive eyes. Ophthalmology. 2008;115:2253-8.
- Karcioglu ZA. Fine needle aspiration biopsy (FNAB) for retinoblastoma. Retina. 2002;22(6):707-10.
- O’Hara BJ, Ehya H, Shields JA, et al. Fine needle aspiration biopsy in pediatric ophthalmic tumors and pseudotumors. Acta Cytol. 1993;37(2):125-30.
- Stevenson KE, Hungerford J, Gardner A. Local extraocular extension following intraocular surgery. Br J Ophthalmol. 1989;73:739-42.
- Palma J, Sasso DF, Dufort G, et al. Successful treatment of metastatic retino-blastoma with high-dose chemotherapy and autologous stem cell rescue in South America. Bone Marrow Transplant. 2011.
- Lucignani G, De Palma D. PET/CT in paediatric oncology: clinical usefulness and dosimetric concerns. Eur J Nucl Med Mol Imaging. 2011;38:179-84.
- Dunkel IJ, Jubran RF, Guruangan S, et al. Trilateral retinoblastoma: potentially curable with intensive chemotherapy. Pediatr Blood Cancer. 2010;54(3):384-7.
- Canturk S, Qaddoumi I, Khetan V, et al. Survival of retinoblastoma in less-developed countries impact of socioeconomic and health-related indicators. Br J Ophthalmol. 2010;94:1432-6.
- Dai S, Dimaras H, Héon E, et al. Trilateral retinoblastoma with pituitary-hypothalamic dysfunction. Ophthalmic Genetics. 2008;29(3):120-5.
- Berry JL, Jubran R, Kim JW, Wong K, Bababeygy SR, Almarzouki H, et al. Long-term outcomes of group D eyes in bilateral retinoblastoma patients treated with chemoreduction and low-dose IMRT salvage. Pediatr Blood Cancer. 2013;60(4):688-93.
- Razek A, Elkhamary S. MRI of retinoblastoma. BJR. 2011;84:775-84.
- Reinoso-Barbero F, Herranz-Ortega MA, Lahoz-Ramón A. Anestesia para cirugía oftálmica pediátrica. En: Actualización en cirugía oftálmica pediátrica. Capítulo 2. LXXVI Ponencia Oficial de la Sociedad Española de Oftalmología. 2000;2:21-31.