Volumen 20 - Número 1 - Enero-Marzo 2012
Manifestaciones oftalmológicas en el déficit de 3-hidroxiacil-CoA deshidrogenasa de cadena larga
J. Català Móra, D. Hajjar Sesé, J. DÃaz Cascajosa, J. Prat Bartomeu
Servicio de Oftalmología.
Hospital Sant Joan de Déu.
Hospital Sant Joan de Déu.
CORRESPONDENCIA
Jaume Català Móra.
Hospital Sant Joan de Déu.
Passeig Sant Joan de Déu, 2 .
08950 Esplugues de Llobregat.
Hospital Sant Joan de Déu.
Passeig Sant Joan de Déu, 2 .
08950 Esplugues de Llobregat.
RESUMEN
Introducción: Definir los hallazgos oftalmológicos del déficit de 3-hidroxiacil-CoA deshidrogenasa de cadena larga.
Material y métodos: Presentación y discusión de un caso clínico de un paciente con este error congénito del metabolismo beta-oxidativo mitocondrial.
Resultados: Paciente de 4 años de edad diagnosticado a los 5 meses de déficit de 3-hidroxiacil-CoA deshidrogenasa de cadena larga, en seguimiento oftalmológico por alteración pigmentaria difusa de la retina.
Conclusión: En este déficit hay un exceso de intermediarios del metabolismo de los ácidos grasos de cadena larga y muy larga que producen retinopatía, por lo que los pacientes deben alimentarse un una dieta especial.
Material y métodos: Presentación y discusión de un caso clínico de un paciente con este error congénito del metabolismo beta-oxidativo mitocondrial.
Resultados: Paciente de 4 años de edad diagnosticado a los 5 meses de déficit de 3-hidroxiacil-CoA deshidrogenasa de cadena larga, en seguimiento oftalmológico por alteración pigmentaria difusa de la retina.
Conclusión: En este déficit hay un exceso de intermediarios del metabolismo de los ácidos grasos de cadena larga y muy larga que producen retinopatía, por lo que los pacientes deben alimentarse un una dieta especial.
RESUM
Objectiu: Definir les troballes oftalmològiques del dèficit de 3-hidroxiacil-CoA
deshidrogenasa de cadena llarga.
Material i mètodes: Presentació y discusió d’un cas clínic d’un pacient amb aquest error congènit del metabolisme beta-oxidatiu mitocondrial.
Resultats: Pacient de 4 anys d’edat diagnosticat als 5 mesos de dèficit de
3-hidroxiacil-CoA deshidrogenasa de cadena llarga, en seguiment
oftalmològic per alteració pigmentària difusa de la retina.
Conclusió: En aquest dèficit hi ha un excés d’intermediaris del metabolisme dels àcids greixosos de cadena llarga i molt llarga que produeixen retinopatia, pel que els pacients han de seguir una dieta especial.
deshidrogenasa de cadena llarga.
Material i mètodes: Presentació y discusió d’un cas clínic d’un pacient amb aquest error congènit del metabolisme beta-oxidatiu mitocondrial.
Resultats: Pacient de 4 anys d’edat diagnosticat als 5 mesos de dèficit de
3-hidroxiacil-CoA deshidrogenasa de cadena llarga, en seguiment
oftalmològic per alteració pigmentària difusa de la retina.
Conclusió: En aquest dèficit hi ha un excés d’intermediaris del metabolisme dels àcids greixosos de cadena llarga i molt llarga que produeixen retinopatia, pel que els pacients han de seguir una dieta especial.
ABSTRACT
Aim: To define the ophthalmologic findings in the deficit of long-chain 3-hydroxy acyl-coenzyme A dehydrogenase (LCHAD).
Material and methods: Presentation and discussion of a case report of a patient with this congenital beta-oxidative mitochondrial metabolism error.
Results: Patient aged 4 diagnosed at 5 months of LCHAD deficit, following ophthalmologic controls by diffuse pigmentary disorder of the retina.
Conclusion: This deficit produces an excess of intermediate long and very long chain fatty acids producing retinopathy, so patients should follow dietary therapy.
Material and methods: Presentation and discussion of a case report of a patient with this congenital beta-oxidative mitochondrial metabolism error.
Results: Patient aged 4 diagnosed at 5 months of LCHAD deficit, following ophthalmologic controls by diffuse pigmentary disorder of the retina.
Conclusion: This deficit produces an excess of intermediate long and very long chain fatty acids producing retinopathy, so patients should follow dietary therapy.
Introducción
El déficit de 3-hidroxiacil-CoA deshidrogenasa de cadena larga (LCHAD por sus siglas en inglés) es un trastorno congénito del metabolismo de los ácidos grasos con herencia autosómica recesiva. Es debido a mutaciones mediante las cuales se produce un acúmulo de intermediarios de este metabolismo, produciéndose tempranamente crisis metabólicas potencialmente letales con hipoglucemia hipocetósica y acidosis; también aparecen cardiomiopatía, hipotonía, hepatomegalia, ictericia, polineuropatía periférica y rabdomiolisis1. Sobre los efectos de la alteración metabólica del feto sobre la madre se ha postulado su relación con la preeclamsia, el Síndrome HELLP, la hiperémesis gravídica, el Síndrome Antifosfolípido y el infarto de base de placenta, aunque aún están por dilucidar los mecanismos2.En el LCHAD la síntesis de DHA (ácido docosahexaenoico) está bloqueada a nivel del ácido alfa-linoleico. Siendo que el DHA es uno de los ácidos grasos de membrana más abundantes en los fotorreceptores, y que existe déficit en algunas patologías retinianas de similares características, se ha postulado en la literatura científica que una suplementación detética podría beneficiar a pacientes con esta enfermedad. Partiendo de esta base ya se han iniciado estudios clínicos con suplementos de DHA en la dieta, además de la dieta propia para la metabolopatía, cuyos resultados iniciales muestran que podría producirse un enlentecimiento o detención de la progresión de la retinopatía3,4.
En la esfera oftalmológica el hallazgo más relevante lo encontramos en el polo posterior3,5,6. Se desarrolla una retinopatía pigmentaria en la cual inicialmente, la retina puede tener un aspecto más claro, pero posteriormente vira hacia una condensación y agregación del pigmento retiniano del polo posterior y la región macular. Otros hallazgos de polo posterior son estafilomas y algún caso de membrana neovascular subretiniana. En cuanto a la afectación de polo anterior hay
documentados casos de opacidades cristalineanas y cataratas7. En las pruebas funcionales encontramos alteración de las amplitudes en el electrorretinograma (ERG), algunos casos de potenciales evocados visuales (PEV) con menores amplitudes, pero sin retraso en la transmisión, y un campo visual con un escotoma paracentral que progresa centrípetamente. Los pacientes presentan asimismo alteraciones en la visión cromática y nictalopia en fases avanzadas de la enfermedad.
Presentamos un caso de un paciente varón de 4 años de edad diagnosticado a los 5 meses después de nacer de déficit de 3-hidroxiacil-CoA deshidrogenasa de cadena larga a raíz de cuadro de ictericia y pérdida de apetito. En la exploración física, además de la ictericia cutáneo mucosa, presentaba coluria, hepatomegalia sin esplenomegalia, leve hipotonía axial y disminución de peso. Los parámetros analíticos mostraban un síndrome colestásico, con inversión del cociente ALT/AST, hiperbilirrubinemia y bilirrubinuria. La ecografía abdominal solo revelaba hematomegalia con vías biliares normales. Exploración de fondo de ojo normal. Se inició despistaje de metabolopatías; el estudio de acilcarnitinas en fibroblastos mediante espectrometría de masas reveló un perfil compatible con de déficit de 3-hidroxiacil-CoA deshidrogenasa de cadena larga. Tras el inicio de dieta baja en ácidos grasos y suplemento de L-carnitina el cuadro del paciente mejoró. El estudio de genética molecular reveló la presencia de dos mutaciones: G1528C, la más frecuente en este trastorno, y G2107A, esta última cuya presencia no está descrita en la literatura.
Durante los controles oftalmológicos el paciente tenía una agudeza visual de 0,8 en ojo derecho y 0,7 en ojo izquierdo sin corrección, test de Lang positivo, y en el fondo de ojo apreció una retinopatía en sal y pimienta periférica junto con un aumento de la pigmentación macular, con árbol vascular normal (Figura 1 y Figura 2). Se realizaron estudios con Electrorretinograma, Potenciales evocados visuales y Tomografía de coherencia óptica, resultando dentro de la normalidad.
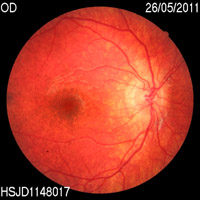
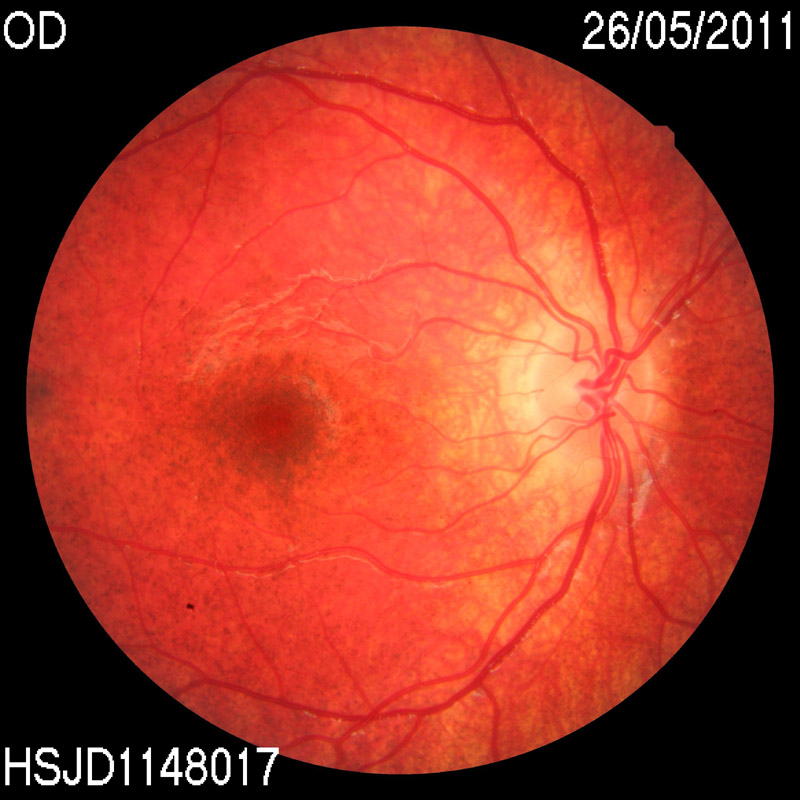
Figura 1
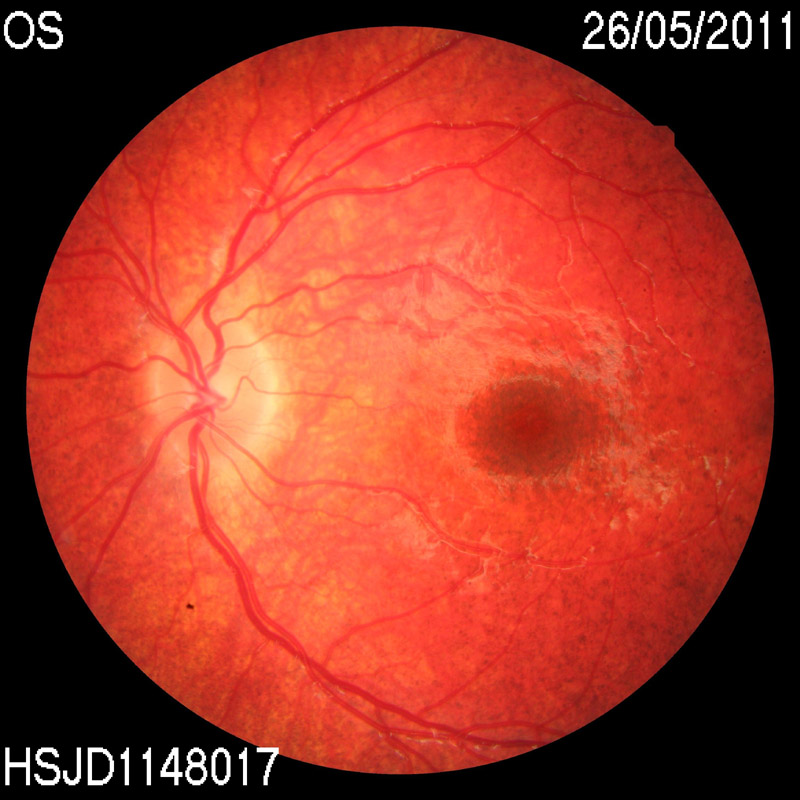
Figura 2
Bibliografía
- Tyni T, et al. Ophthalmologic findings in long-chain 3-hidroxyacyl-CoA dehidrogenase deficiency caused by the G1528C mutation. Ophthalmology 1998; 105:810-24.
- Rakheja D, et al. Long-Chain L-3-Hydroxyacyl-Coenzyme A Dehydrogenase Deficiency: A Molecular and Biochemical Review. Lab Invest 2002, 82:815-24.
- Gillingham MB, et al. Effect of optimal dietary therapy upon visual function in children with long-chain 3-hydroxyacyl CoA dehydrogenase and trifunctional protein deficiency. Mol Genet Metab. 2005 Sep-Oct;86(1-2):124-33.
- Fahnehjelm KT, et al. Ocular characteristics in 10 children with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: a cross-sectional study with long-term follow-up. Acta Ophthalmol Scand 2008;86:329-37.
- Sturm V. Ophthalmologic abnormalities in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: presentation of a long-term survivor. Eur J Ophthalmol. 2008 May-Jun;18(3):476-8.
- Schrijver-Wieling. Retinal dystrophy in long chain 3-hydroxy-acyl-coA dehydrogenase deficiency; British Journal of Ophthalmology 1997;81:291-4.
- Russell-Eggitt et al; Cataract in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD). Ophthalmic Genet. 2003 Mar;24(1):49-57.